 AUTO-MOL-FIT AUTO-MOL-FIT
AUTO-MOL-FIT AUTO-MOL-FITBoth molecules should be chemically equivalent (i.e. equal number of atoms, the correct atom type assigned, no hydrogen atoms missing).
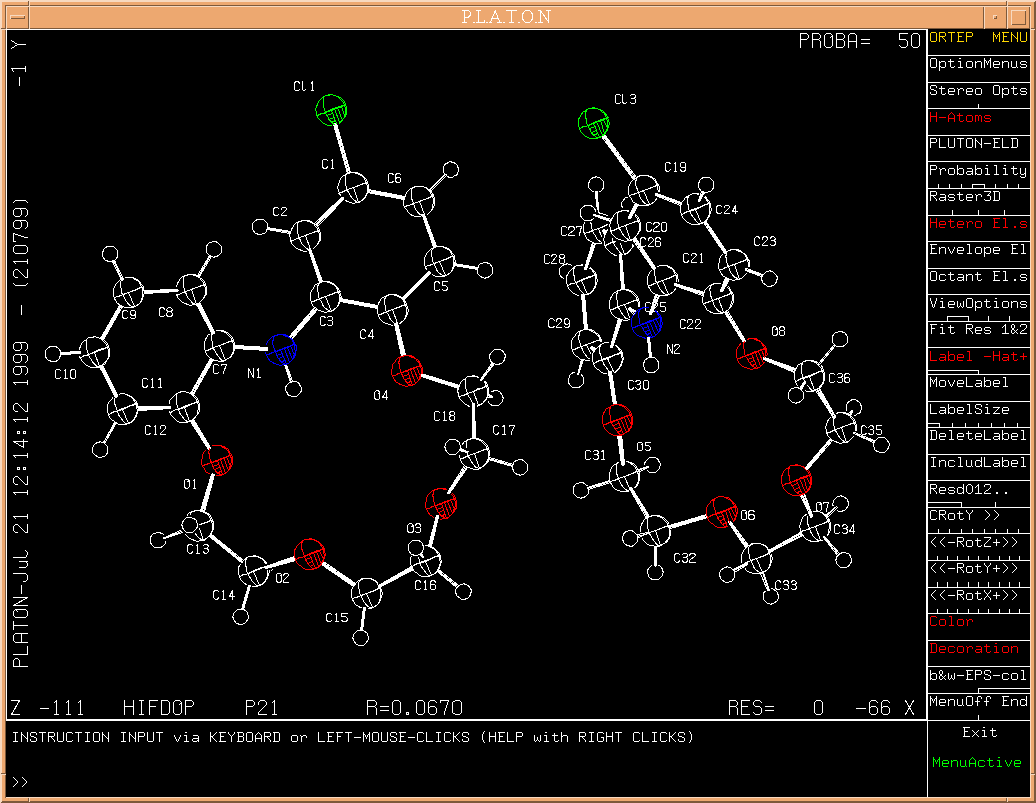
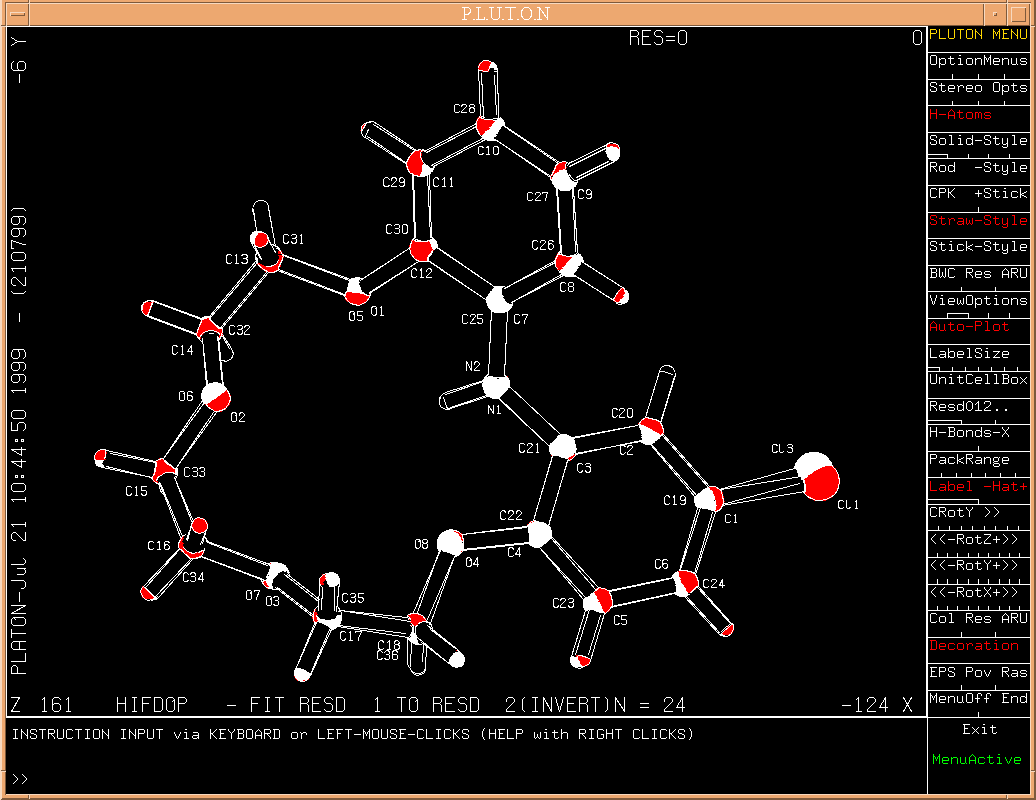
Example: A structure taken from the CSD in P21, Z = 4. [associated data]. AutoMolFit result.
The automatic fitting algorithm involves unique numbers based on network topology assigned to all atoms in the structure. Topology numbers are listed in the connectivity table under the heading 'tnr'.
Automatic fitting is attempted, using the quaternion fit technique, ( A.L. Mackay, Acta Cryst.(1984), A40, 165-166), on the basis of atoms with a unique and equivalent topology number in each molecule to be fitted. Atoms that are not topologically unique in a molecule are not included in the fit calculation (but shown in the subsequent plot).
The published Mackay procedure fails for (close to) 180 degree fit rotations about an axis. The 180 degree situation is of course quite common in the crystallographic setting. PLATON/FIT implements a special 'work-around' for this problem.
The fit is done of the first residue or its inverted image on the second residue: the best fit is retained and displayed (along with the number of atoms on which the fit was done).
By default (i.e. without the specification of residue numbers), residue #1 is fitted on residue #2. Other fits should be specified explicitly from the keyboard (e.g. 'FIT 2 3').
Hydrogen atoms are not included in the automatic fit, but included in the subsequent PLUTON style display.
Details on the fitting results are written to the listing files.
In the case that the molecules to be fitted have no or not enough unique atom pairs, fitting is attempted assuming consistent atom numbering in both molecules.
The ORTEP function provides an option to click on a set of atom pairs on which the fitting should be based .
The Quaternion fit algorithm is also part of the NONSYM function. Assignment of equivalent (corresponding) atoms is done differently there. Symmetrical molecules may often be fitted automatically via that path.
A fit on an explicit subset (at least 5 pairs) of atoms can be done
as well:
Example: 'FIT N1 N2 O1 O5 O2 O6 O3 O7 O4 O8'
The fitted coordinate sets are written to a file 'compound_fit.spf'.
See other FIT-Options
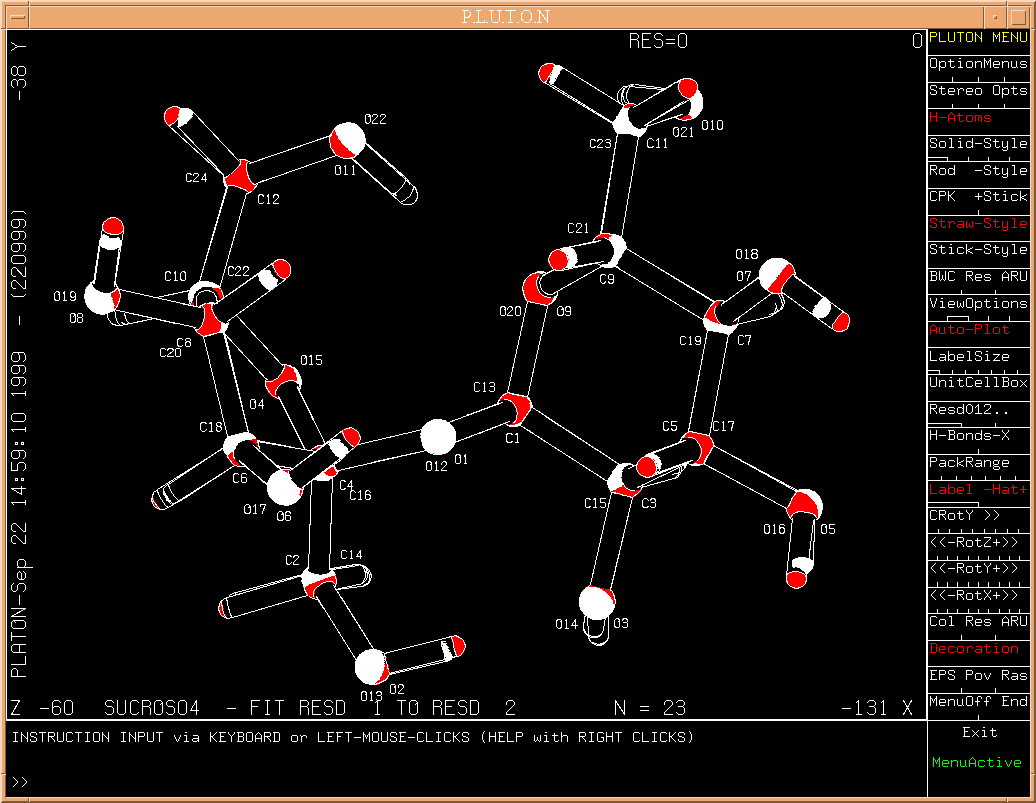
The number of atoms in both molecules may differ.
Example: A fit of the structure of sucrose from neutron data on
that of X-rays
[data and
fit-result].
 PLATON HOMEPAGE
PLATON HOMEPAGE
{kind=link}
{kind=link}
{kind=link}